Genomic contingencies in evolution
The majority of my research focuses on how genomes themselves evolve and in turn how their structure influences evolution at the organismal level. Processes and phenomena like the efficacy of natural selection, the spread of selfish genetic elements, and the leakiness of species boundaries are influenced at the most basic level by structural traits like linkage between loci, where and how often chromosomes recombine, and the fitness consequences of rearranging local structure. Genomes themselves are in constant flux, undergoing point and structural mutation, translocating parts of chromosomes, doubling themselves in whole or in part, and fighting back and corralling selfish elements. I’m fascinated by this dynamic genomic world and how evolution at this level can influence the fitness and evolution of the organism that same genome encodes. In much of my doctoral work, I’ve made use of major transitions in ploidy and mating system, where we may expect changes in fundamental traits like rates of mutation and recombination, the distribution of fitness effects, and the efficacy of selection to change the rules of evolution, to address some of the questions below in a comparative framework. In my postdoc, I’m exploring the impacts of demography and selection on genetic diversity and the efficacy of selection in Aedes. Below I briefly cover some of the topics I’ve chosen to pursue under this general interest of genomic structure and contingency in evolution.
Recombination Evolution

Meiotic recombination is a major population genetic parameter that determines the evolutionary dynamics in genomes, and has been described as the primary source shaping evolution (Stebbins 1950). Recombination tends to localize to certain genomic regions, which are often upstream of genes in regions of open chromatin in plants, where the average recombination rate is many times higher than the background rate. Despite being a fundmental source of variation, we know surprisingly little, especially in plants, about how and why rates of recombination vary within and between species. Additionally, recombination rates have been theorized to evolve differently in species with contrasting mating systems, and the genomic landscape of recombination may evolve in response to other genomic features like transposable elements. I’m interested in how recombination rates evolve and the factors that determine their genomic distribution within and among species, and how the recombination landscapes of genomes determine how evolution proceeds.
Relevant papers: Kent, Uzunović, and Wright 2017 Phil Trans Roy Soc B
Introgression
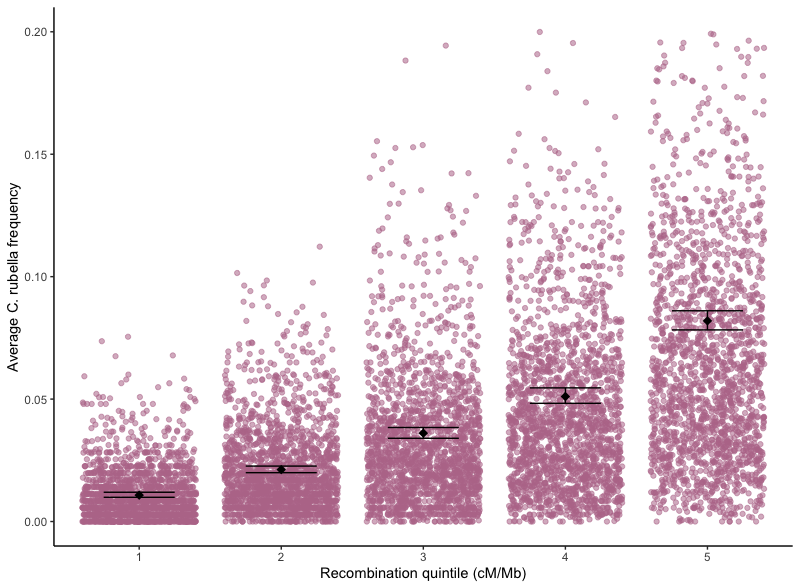
I am generally interested in how gene flow and introgression influence and prevent adaptation, and in what shapes the genomic landscape of admixed ancestry. Admixed ancestry has the potential to be positively selected and influence the adaptation of species or act as deleterious mutational input that reinforces species boundaries. These opposing forces of selection, in combination with normally-occuring genomic processes like background selection can create similar patterns that are difficult to distinguish in genomic data. Additionally, what determines where in the genome gene flow can leak through species boundaries and persist is dependent on the genomic architecture of selected sites and their correlation with recombination at fine and broad scales in complex and fascinating ways. In my work, I aim to untangle the roles of recombination, selection, and genome structure in driving patterns of introgression using empirical data and simulations. I have worked on projects investigating the role of introgression in evolution in selfing and outcrossing Capsella species pairs, diploid and polyploid Capsella species pairs, and wild and domesticated Oryza species pairs.
Relevant papers: Sas et al. 2016 Current Biology
Kryvokhyzha and Salcedo et al 2018 PLoS Genetics
Linked Selection

When genomic sites undergo positive or negative selection, the neutral sites surrounding them experience linked selection (known as selective sweeps and background selection, respectively). This results in a loss of neutral diversity near selected sites, with impacts depending on the strength of selection and the local recombination rate. Although many studies have investigated either selective sweeps or background selection, it has been difficult to investigate their joint impacts. Additionally, in species with reduced recombination or efficacy of selection, the effects of linked selection can become more complicated, as selected sites begin to interfere with each other, and in polyploids, genomic redundancy could lead to lower or higher impacts of linked selection. I’m interested in parameterizing the joint effects of linked selection in a comparative context, to better understand how the diversity of genome size and structure alters our expectations.